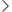技术文章
Technical articles 更新时间:2026-01-23
更新时间:2026-01-23 点击次数:384
点击次数:384
肿瘤细胞远端转移后常进入“休眠",但它们如何在免疫监视与组织微环境压力下维持存活、并在未来某个时点重新爆发,机制仍不清晰。尤其是 EMT 并非单一状态:不同形式 EMT 的“力学表型"与免疫逃逸之间是否存在因果链条,一直缺少直接机制证据。
该研究显示:在肺腺癌(LUAD)休眠转移模型中,TGFβ 诱导的 EMT 会从“典型全 EMT(应力纤维丰富、迁移性强)"进一步转变为一种“非典型 EMT(细胞变圆、缺乏应力纤维、皮质肌动蛋白为主)";这一转变由肌动蛋白去聚合蛋白 gelsolin(GSN)驱动,导致细胞生物力学刚度下降,从而降低被 NK/CTL 等细胞毒性淋巴细胞“机械监视/力学杀伤"的概率,帮助休眠细胞在免疫监视下存活。

结论 1:休眠转移细胞存在持续的 TGF-β 信号活性,且 TGF-β 对“潜伏后仍具转移爆发能力"的维持至关重要
作者在休眠播散细胞中观察到持续的 TGF-β 报告基因活性并伴随低 Ki67 等静息表型;更关键的是,在免疫细胞“早期清除"与“晚期解除监视(模拟潜伏后免疫松动)"两种情境下,Tgfbr2 敲除对晚期爆发的抑制更显著:晚期去除 NK/CD4/CD8 后,Tgfbr2-KO 组转移爆发显著减少,提示 TGFβ 信号并非只是“进入休眠"的触发器,更像是“维持可存活/可再起"的必要条件之一。

Fig1. 休眠LUAD细胞中的重要TGFβ信号传导
结论 2:持续 TGF-β 刺激诱导“先典型 EMT、后非典型 EMT"的时间序列,并与增殖静息耦合
在 dormancy-competent 的 LUAD 祖细胞模型(如 LCC)中,TGF-β 早期诱导典型 EMT:细胞拉长、上应力纤维、迁移增强;但随着刺激持续,细胞逐渐变圆、应力纤维消失、运动性下降,同时 EMT 转录程序仍然上调(“形态/力学"与“转录 EMT"出现解耦),并伴随 Ki67 下降、p27 等静息标志上升。
Fig2. 静止相关EMT对TGF-β的反应
结论 3:GSN 是 TGF-β 驱动“非典型 EMT + 静息"联动的关键枢纽,且其上调与增强子乙酰化及单细胞轨迹一致
作者从 RNA-seq 中锁定 GSN 等细胞骨架调控基因,并验证 GSN mRNA/蛋白在 TGF-β 持续刺激下逐步上升;ChIP-seq/ChIP-PCR 指向 GSN 位点增强子 H3K27ac 升高;单细胞转录组时间序列显示:GSN 高表达细胞往往同时具有更高 EMT 得分与更强静息特征,提示 GSN 并非旁观者,而是贯穿状态转变的“轴心变量"。

Fig3. Gelsolin与静止的EMT结合
结论 4:GSN 介导的肌动蛋白重塑降低细胞刚度,从而削弱 NK/CTL 介导的“机械监视式杀伤",并在体内决定休眠细胞能否逃避免疫监视
功能上,GSN 敲低不影响早期“拉长"的诱导,但会阻断后期向“变圆/皮质肌动蛋白为主"的转变;力学层面,作者用力学测量显示 GSN 敲低提高细胞刚度;免疫效应层面,TGF-β 处理 7 天(更偏“非典型 EMT"状态)的细胞相较 3 天更耐 CTL 杀伤且引发更弱的 CTL 激活;而 GSN 缺失细胞虽然可以进入休眠,但更容易在免疫监视下被清除。作者还通过“直接操控肌动蛋白状态"的方式(如 DeAct)对免疫敏感性进行回证,强化了“骨架→刚度→免疫杀伤概率"的因果链。

Fig. 4 细胞刚度降低所致的免疫逃逸表型
研究逻辑总结:
1)先在体内休眠转移模型中确认:潜伏的转移细胞呈现持续 TGF-β 活性与静息特征,并用 Tgfbr2-KO + “早/晚期免疫去除"实验将 TGF-β 的作用从“进入休眠"推进到“维持潜伏爆发能力"。
2)再把关键现象在体外重建为时间序列:TGF-β 诱导典型 EMT → 进一步转入缺乏应力纤维的非典型 EMT,并伴随细胞周期停滞。
3)随后用组学锁定并验证 GSN:TGF-β 诱导其表达与增强子活化,单细胞测序显示 GSN 是 EMT 与静息共同演化的关键。
4)把“骨架重塑"落到“力学表型"与“免疫杀伤结局":刚度下降带来 NK/CTL 杀伤逃逸;GSN 缺失则提高刚度、提高被杀概率,并在体内决定休眠细胞存亡。

 当前位置:
当前位置: